Download Script
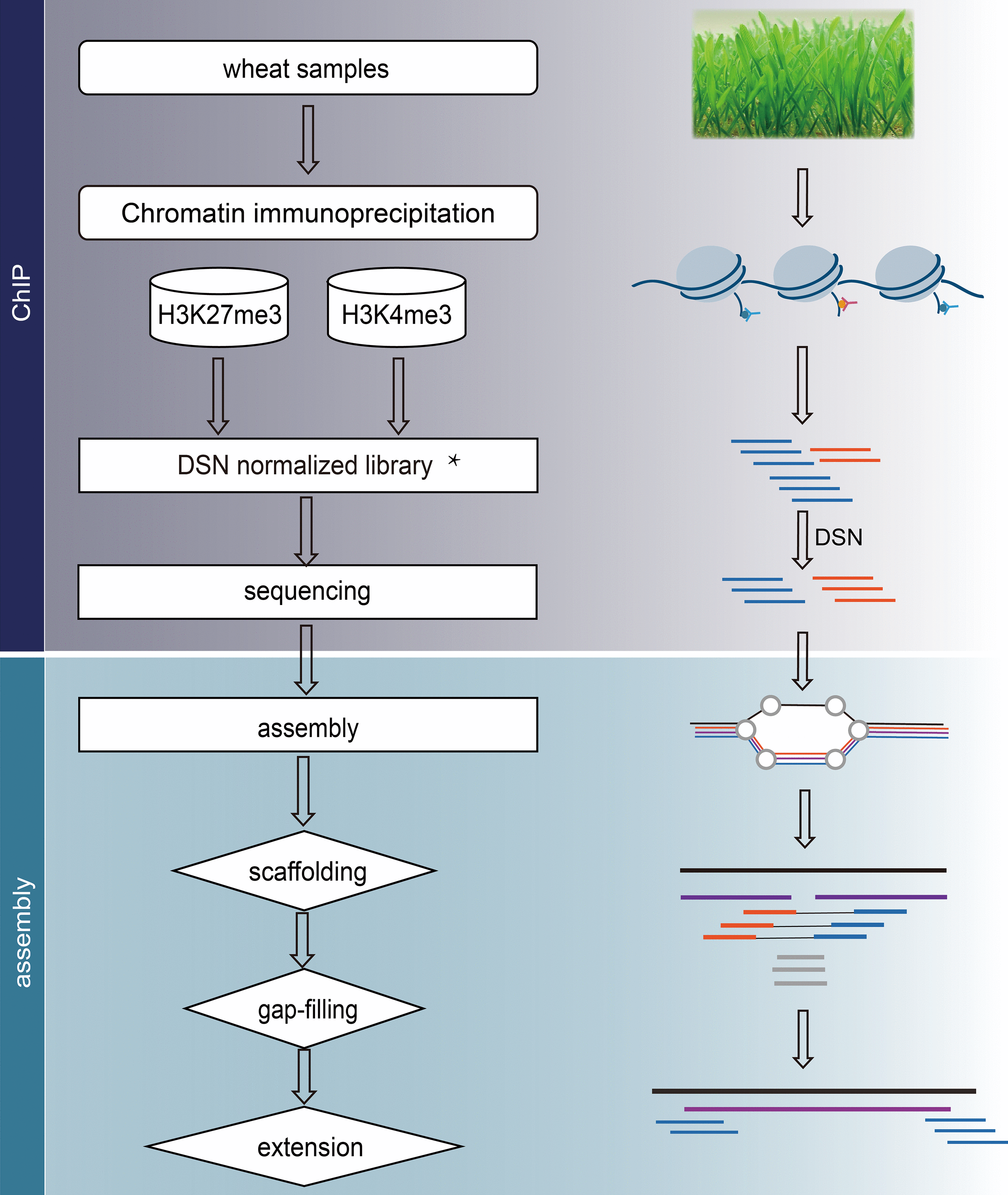
cgt-seq.shIntroduction
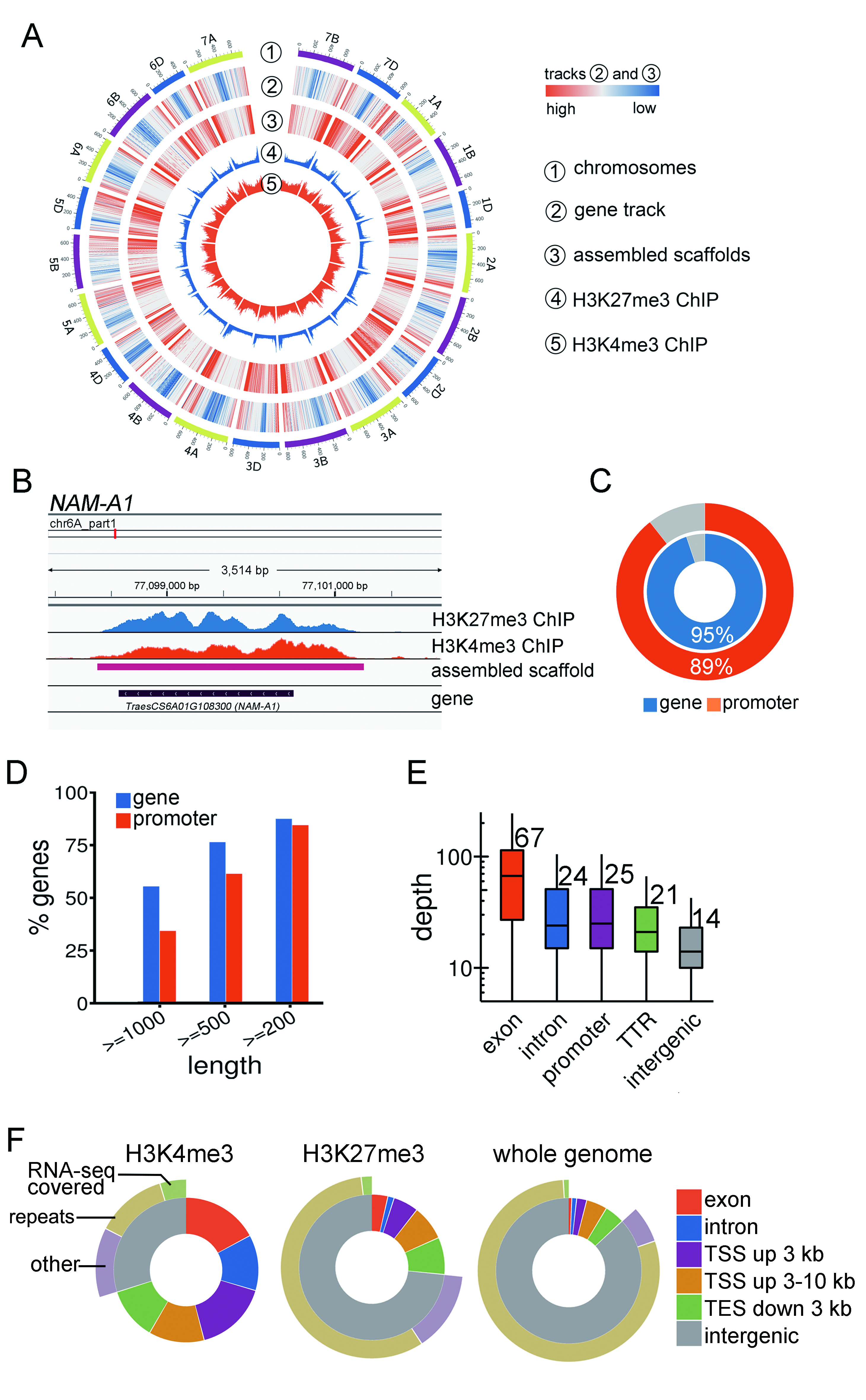
Genetic diversity in plants is remarkably high. Recent whole genome sequencing (WGS) of 67 rice accessions recovered 10,872 novel genes. Comparison of the genetic architecture among divergent populations or between crops and wild relatives is essential for obtaining functional components determining crucial traits. However, many major crops have gigabase-scale genomes, which are not well-suited to WGS. Existing cost-effective sequencing approaches including re-sequencing, exome-sequencing and restriction enzyme-based methods all have difficulty in obtaining long novel genomic sequences from highly divergent population with large genome size. The present study presented a reference-independent core genome targeted sequencing approach, CGT-seq, which employed epigenomic information from both active and repressive epigenetic marks to guide the assembly of the core genome mainly composed of promoter and intragenic regions. This method was relatively easily implemented, and displayed high sensitivity and specificity for capturing the core genome of bread wheat. 95% intragenic and 89% promoter region from wheat were covered by CGT-seq read. We further demonstrated in rice that CGT-seq captured hundreds of novel genes and regulatory sequences from a previously unsequenced ecotype. Together, with specific enrichment and sequencing of regions within and nearby genes, CGT-seq is a time- and resource-effective approach to profiling functionally relevant regions in sequenced and non-sequenced populations with large genomes.
Background
This approach is inspired by the fact that promoter and gene body regions are enriched for specific types of epigenetic marks. H3K4me3 is the typical active mark and H3K27me3 is the typical repressive mark.
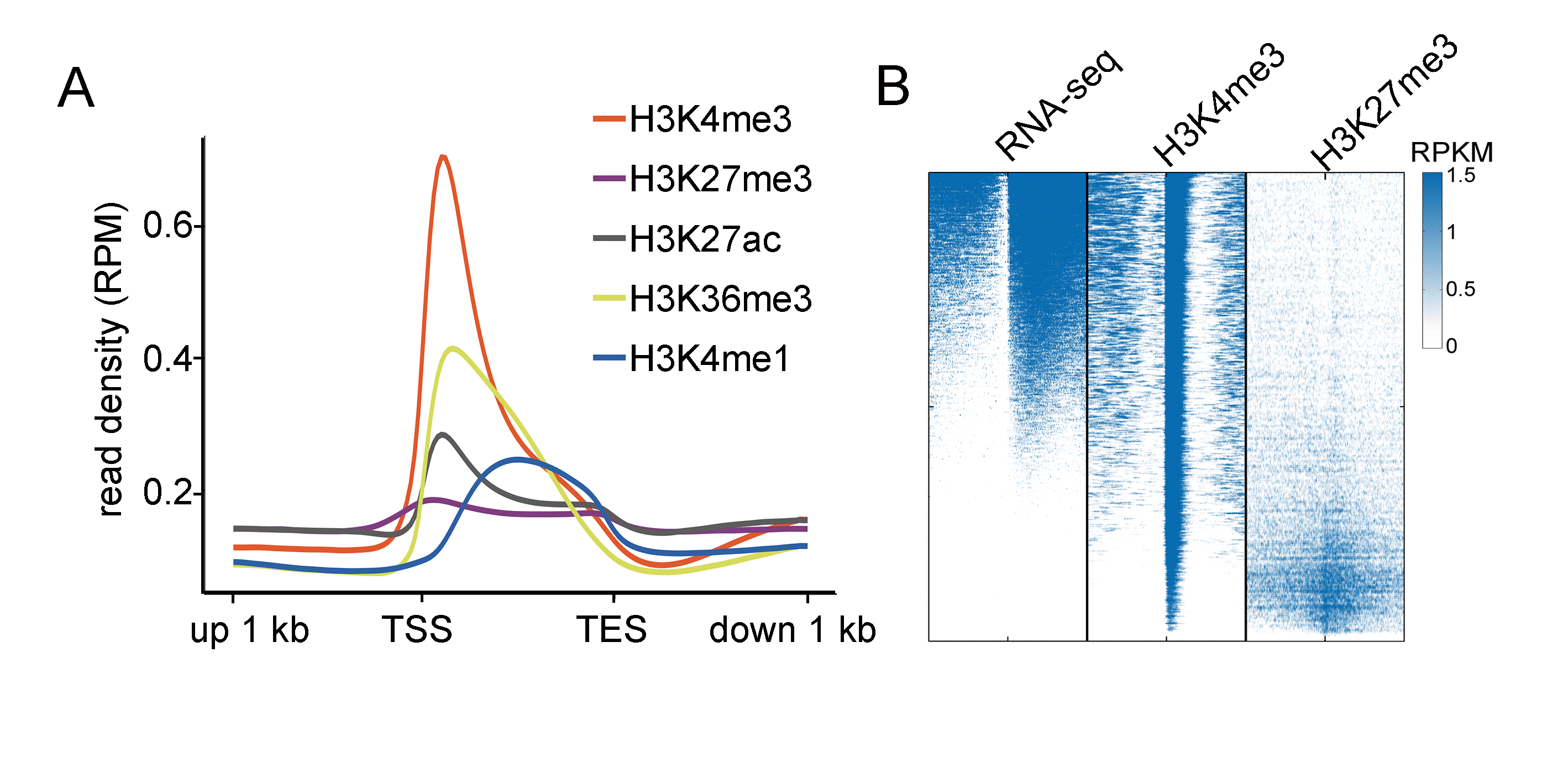
Workflow
Performance
Genome Browsersensitivity and specificity
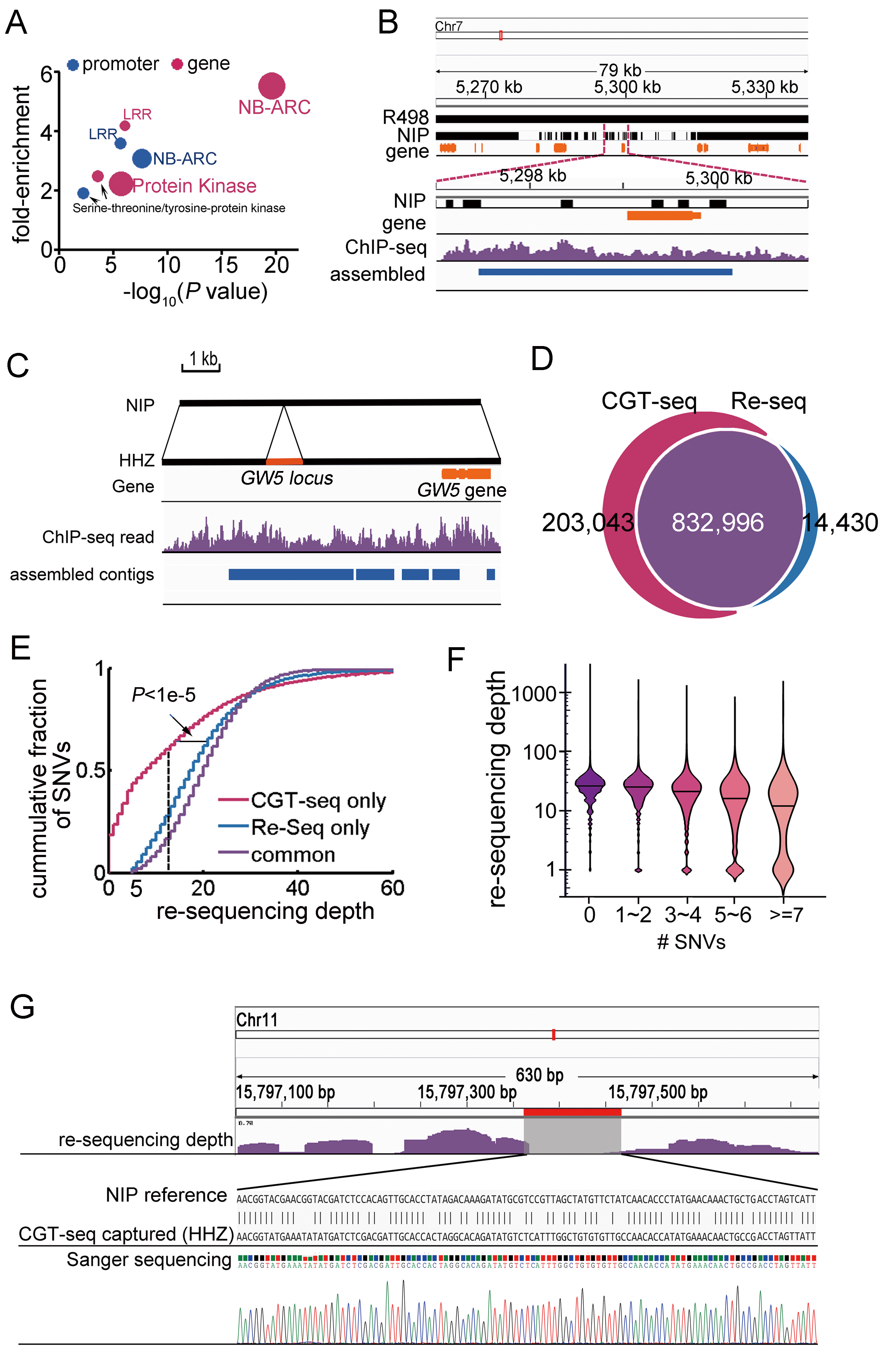
Application
High accurate detection of genic and regulatory sequences and SNVs
Acknowledgements
We thank Prof. Hongxuan Lin from Shanghai Institute of Plant Physiology and Ecology for providing rice materials and re-sequencing data. We thank Prof. Jiankang Zhu from Shanghai Center for Plant Stress Biology for the thoughtful discussions.